|
Breaking-Cas User Guide |
Breaking-Cas is a versatile system for detecting putative sgRNA off-targets in CRISPR/Cas applications. Its main features are summarized in Table 1:
| Tool | Breaking-Cas |
| Website | https://bioinfogp.cnb.csic.es/tools/breakingcas |
| Input | One or several FASTA sequences up to 20,000 nucleotides in total. File uploading allowed. |
| Output | Rich interactive web page containing detailed information about candidate oligos, on-targets and off-targets. Scores, coordinates and overlapping genes are shown. Mini genome browsers allow checking the genomic environment of each putative off-target. Results can be downloaded as tables. |
| Throughput | Medium/High. Multiple queries in batch are allowed. |
| Use cases | To design sgRNAs and evaluate putative undesired off-targets for CRISPR/Cas applications. It can be used for any eukaryotic genome available at ENSEMBL/ENSEMBLGENOMES. |
| Genomes | More than 650 (as of April, 2016). |
| sgRNA constrains | Versatile system: oligo size, mismatch number, PAM sequence and position, and scoring system can be customized by the user. |
| Validation | N/A |
| Pros | Very fast and easy to use. Results are interactive, very detailed and well organized. |
| Cons | Does not include any method to measure sgRNA efficiency. |
| Software | Web |
Table 1. Breaking-Cas's features. See Table 2 of Graham and Root, 2015 for a recent comparison of the very same fields for other similar tools.
This guide illustrates the use of Breaking-Cas with the analysis of the sms-2 gene from Caenorhabditis elegans (roundworm).
Following steps 1 to 3 is equivalent to using the "Fill with example" link at the bottom of Breaking-Cas form.
Step 1. At the input form, select Caenorhabditis elegans as organism (write the name or select it from the alphabetic list).
Step 2. Copy/Paste the nucleotide sequence of gene sms-2 (in FASTA format) in the text area:
>sms-2
GGAGGAAAAATTATGAGTTTCAAAGGGGGAGAAATCGAAAGAAATGAGCAGGAGGGGCATTTTTGCGGGAAAGAAAATGTGTTGTTCGTTGTGTTTAATTGAAATATCTCGTATGGAGCATCGTATTGTACTGGAAAAGAAGTAGGAAAAACCGCTTT
GGGGGAAATACAAAACAAAACATTAACAACAAAAAAATGAGATAAGCAAAATGTCGAAACTGAGGGGAAGGGATTAAATTTTGATTTGAAATTAAACCAAAACTGGGATAAAATAAAATGATACAAAATAAAAAAAAGAGCATTAAATTAAATTAAAA
GTAATACATCAAATTATTGCAATTTGTAGTTGATACGATTCATAATGGTGTGCATTCTTTGAGGGCCTTCCAGCGGCCAATTCCACTTGTTCACCAATTTCCCGTCCGCAACATCCGATTCAAACCAGTAGCACAACCAGAACCACCTGAAAAAAAAT
TTTTTGAATTTTTGATAATAATTTTTCGCGAAAGTTGATATTTGAAGACAATTCTATTTTGGGCACAAGTTTGTTCGCAATAAGAAAATTTTCTCAATGTGCTTTACCCATTTACCAGTTTCTTTGCGTGAATATCATTAACTTGGCCAGTGCAGTTT
ACCACCAATTCGGTCAGTGCATCTTACCAATTTGGTCAGTGCATCTTACCAATTTGGTCAGTGCATCTTACCAATTCGGTCAGTGCATCTTACCAATTTGGTCAGTTTACATAGCTTATTATGTACAAATACTTCCAATTACCTTCTTTCAGTAATTT
TCTACAACTTTTTCTCCCTAATTATCAACTGTTTTCAATTCATTTCCTCACAATCAGAAGCTCTTAACTAACCAAAGTCGACTCAACGGCGCCTGTGGTCGATCGTCCTTTCGCATTTCGAAAATTTGGTGATAAGACCAGAAAACGTGACTTGTCAA
CCAGTACGCAATTAACACGTCCATCGTATAGTGACCGCCTGACACGACGAGAGCTGCGATTCCGAGGAATGTGATCGGAGCGGCAATGTATCTAAAATACAAAAATGAAAAAAATGAATAATGTTTCTCAAAAAAGTTTTTCATTCAAAAAGAATCAC
CTGAGAATAACGAGACCACGCGGAGTGTACTGCAACTGCACAAAATACATAATTGTGAGAACGACAGTGTGCCCGCTAAACATCAGATCACCGCACAGAATCTTGTCTTGTCCGGATGTCAAGCCCAAAGTGATGACGTAGGTGAGGAATCTGAAATA
TGTTTGAATTCAGTTCCAGGAATATTTTTATATCGCAACTTTAAAAAATAGGGCATTTTCCAAACGTTTGCCTGAATATTCTATATGTATATCTAAATTTTTTGGTACGTTTAATCATGATGTTTGGTCACCCTGCACTTTAATAAGTGTATTCACAC
AATAAATCCGTTGGTGCTACTCAGTAGTGGCATATTTTCAGAAATGTGCCCAATGTAAGCAGAATGTAAAAAACGGCATTAATTTTAAGTCATTGAAATAATTTTTAACCCTTAAATTTTATTACTTCCGTAAAATGGCAAAAAGTAGTTTTGCTCAC
AGATGCCGGTTTTTATGAATTTTCCAAATTTCCCTTTCAGATCTTCAATTAATATTTCTCTCAAAAAACTTCCAATTTCGATTCGCAGTTCGAATCAAGCTTTTCAGTTCTCCCAATTTTTCCCACAACCTTTATTATTTTTTCGATTGTTACCTCGT
CGCGATTTCCATGCCGTACATAGCGGTTCTGTTGACTTGTGGCTGGCAAATTTCATCCCTATTGTGAAAAGAGGGCGGCAGGAAAGTGACACCGAGAATCACGGCACGCAGGCCGTACATGATAGCTCCCAAGAGGAATGTGCGGCGGAGCACAATCC
ACCGCTGATGGTGAAGGAAGATGATGGTGAAGGCGACGACGGAGCTGAAAAACAAAAATAAAAATTTGGGAGTTTGGCCGTGGTTTATCATATATGCACAGATTATTCTTTATAGGTCATTAACGTATTTCTATCTCAATGGTTTTTTTTTTCAAATA
TCATTAAAAATCAAACAACTTCATTTTTCTTAAACTTTTTTTCACATGCCCCATGATTGTTTTATGATTTTTACACTTTCACTGCGTTTTATGACTTCTGGTTTTTTCGGTCAGTGCTCTAATCGATTATTTGCATAGTATGGTAGGCTTTTTCTAAA
AATATATTTTAAATACAGATTTCTCAGCAAATAACATTTAAGCCTTGCATTTTTATGACTCTTGTATAGAAATCAAATTCGTTCATCTTGAAGAGACAATAATGAATACGAAAGAATAGTGGTTGTAGTTAAAACCAAATGTGAACTGAATTTCTGAA
CTTATGATTACCACTAGTGCCAAGGTTTATCCAATTATAATGTTAACTTTGTTTCTGACTAGCCTTAAACAAAACGGAACTTTTTCTAGTGCCTACATAATTTCTTCTATATGAAATTCTTCCAACAATTTTTTTTGTCTTTTCAAAATCACACAGAA
ACAGTTAGCTTCTAGGTAAGACCTAATTGCAAAATAGCTTACCTAACAGTCGAGAGCACATCTCCAACACTCCACGCCCATCTTTGTTGTGGAATGATCATGAAAGTGAGATCGGGAAGCGGCTGTCTAGGTACCACGTCGTGAATGACTGTGAGCAA
GAAGAAGTTGAGAAACGCTGATAGCATGAGACACAAAAACGCCGTCAGTGTCTTGAAGCCCTCAGAGTTTCCGTGGTGCTCATGGTGGAATGTGTCTTCGCAGGTGAACTGTAATATTTAATTTGTAATTTTATTAGGCATTTGTTGATATTATTTTT
GTACCCTATTTTTTTGGCAGCTTAAATGCTTTTCCTTCCTTATTTTAGAGCCACTAGTAAAACTATTTTGCATGTGTGTGGTTTTTTTCTGGTTTTTTATCGAAACTAATTGCCAAAGCCTTTTTTTTATTCCTCCATCTTCACGTCAAAAATTTGGC
CCCGTTATAGTTGCAGCTGTGAAACTGATGCCCACGCACTTTCACTCATGCGGAATTATAATATTTCGTCAATTTCAAATTTCGTTCTCAAGTTTAACGAATAGCCGGAATGAATCACCACGCAAGTACGTGGAAAATCAGGCCAATTTCTCTCAGTT
TCTAAAAGTCATATGCCGTCATGGCAGTGCACCTGGCTTTTTTCACACTCCAATGACGCTTTATTCACCGTTTGACGAGTGTTTTAAAGTTTCAAAAGCAGGCTACATTTTTACAAGTTTTCAAAATATTCAAGAGGCACATTTGTATACTGTAAGTT
CCCAGCCTTTATAATGGAGATCTATAACTTGTGAAAATCTAACTTTTGAGACGTTTCTCTTTTCAGTTGGTTTTAGTGTTTTTGTCTGTCTGAGACTTGCATTTTGTGGTATAAAAACCTTTCTGTTAGGCAGTTCCATATATTTTGTTGCCTACTTT
TGATTTTGTCAGTGCCCTGCACTAAGTTTGCCAAAAATCAAGTGTTTTATAACTTTTCGGTAGTTTATTTTTTTATACTACAACTAAATGGCTTAAAAATTTTTTTTGAACCTTTTTCATTCCACGTGATTAGTTTTGTTGTTTTTGGGTCCTAGAGG
TAAGCTGCGCAGCAATTTCCAAAATTTCCAAAATCCGGATCTTCCAGAAACCGAAAACTGTCGAAGTTTTTGGCAAATGTCAAAATTTTCGATTGCCGTTCATTTCAGCCACCGGCATTTTGCAAAATTTTGGATTATTTAGATACATGGTATAAATC
CAAAATTTCTCAAAGTATTTCCTAATTTCCACTAACCTCTTTCCTAATTGGTGTTGGCTCCGGATCAATTGGGTCTATATTGATAACTATCCCATTACTAACACATGGGTCCCGTGACTGGAGCACATCTGTGAACTCCGAACTGTTTGTCATCT
(Alternatively, upload a text file containing the same sequence).
Step 3. Select settings for the nuclease of interest (Streptococcus pyogenes Cas9 by default) or introduce your own parameters:
Positional weights are editable to take into account position-dependent relevance of mismatches in the oligo when calculating off-target scores (see Box 1 below). A value of "0" means that the mismatch is 100% allowed by the nuclease whereas a value of "1" means that any off-target with a mismatch in this position will not be recognized by the nuclease (Soff=0). Weights have been obtained experimentally by evaluating the nuclease activity in an array of oligos covering a large space of the sequence positilities (e.g. those obtained by Hsu et al (2013) for Cas9, included in this server in the presets for that nuclease). Changing these values (as well as the other parameters of the nuclease) allows to use this server with generic experimental datasets on nuclease preferences (e.g. unpublished) provided they are compatible with Hsu's formulation. Nevertheless, this is an advanced feature and should be used only by experienced users. Wrong weights can lead to misleading off-target scores. Inexperienced users are advised to leave the default values offered by the server.
Finally, introduce your email to receive a message when the analysis is finished (optional). and click Submit button.

If all input fields are correct, a "waiting room" page will open and the analysis will initiate. Depending on the size of input sequences and genome, the process will take from a few seconds to several hours.
When the analysis is finished, the results can be opened on a new window (available on-line for a few days) or downloaded as a compressed folder (recommended). In this last case, uncompress this file and open "index.html" with any current web browser to see the interactive results page.
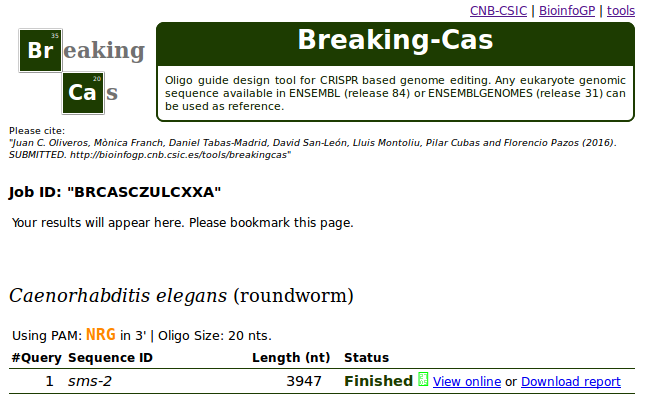
Interactive Results Page
Oligo candidate details and their genomic targets are presented as an interactive web page divided in three main parts: Upper Section, Left Panel and Right Panel.
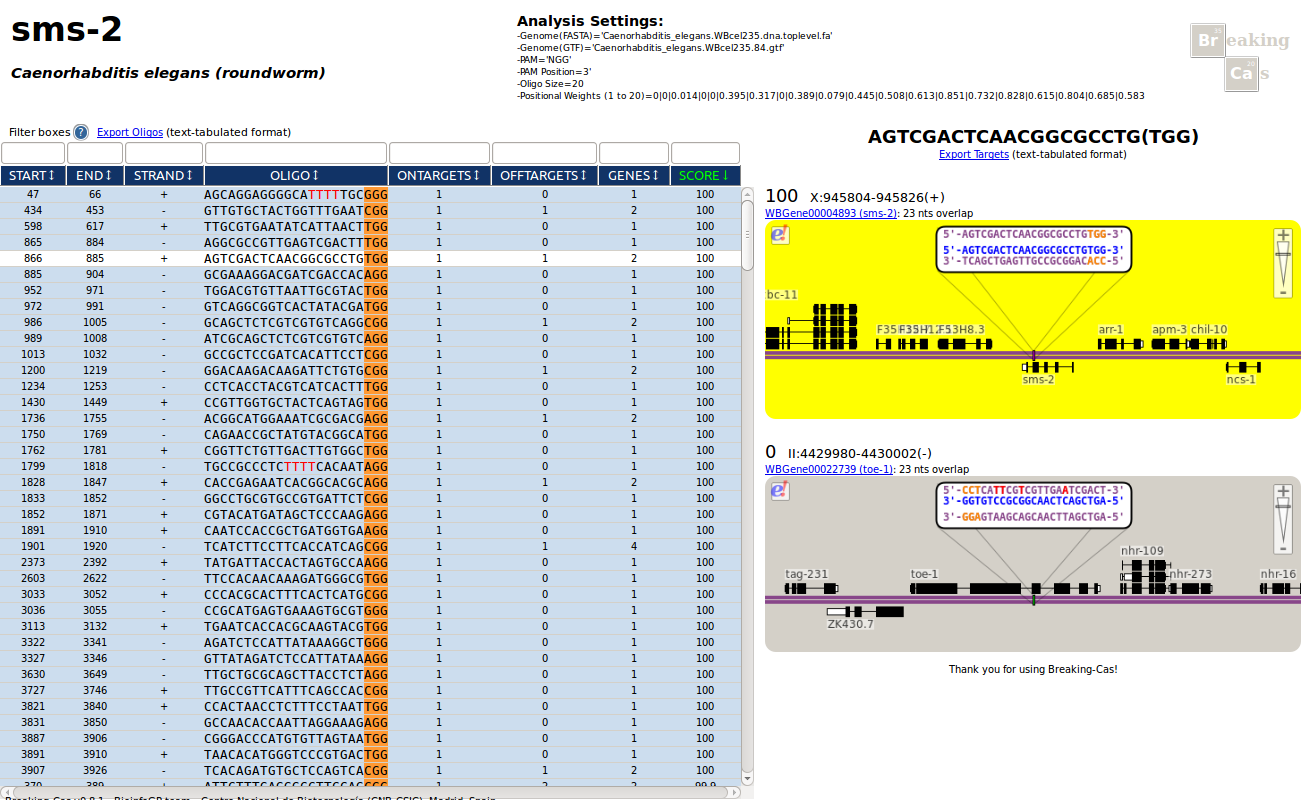
UPPER SECTION: Project Details
 |
|
|
Input sequence ID, Organism name, and parameters used are indicated. |
LEFT PANEL: Oligo Candidates
This table shows all oligo candidates found for the input sequence
(e.g. 20 nt followed by a compatible PAM (NRG) in the case of Cas9).
By default results sorted by the aggregated score.
Thanks to the explicit PAM sequence provided, users can limit their gRNA candidates to those with a desired PAM
while allowing a more generic PAM for off-targets. The typical case is S. pyogenes's Cas9, where NGG is desirable for
gRNAs, whereas off-targets can contain NAG or NGG (NRG in IUPAC).

|
|
Left panel details:
| Caption | Description |
| START | Start position of the oligo in the input sequence. |
| END | End position of the oligo in the input sequence. |
| STRAND | Strand (+ or -) of the input sequence were the oligo was found. |
| OLIGO | Nucleotide sequence of the oligo candidate. PAM is also indicated. |
| ONTARGETS | Number of alignments with zero mistmatches in the reference genome. |
| OFFTARGETS | Number of alignments with one or more mistmatches in the reference genome. |
| GENES | Number of overlapping or nearby genes. Both on-targets and off-targets are considered. |
| SCORE | Aggregated score (Sguide) based on the number and quality of off-targets (see BOX 1 for details) |
RIGHT PANEL: Target Details of Selected Oligo Candidate
When an oligo is clicked on the left panel, target details will appear in the form of minibrowsers illustrating the hybridization sites in the genome.
|
|

|
Comparative Example
A comparison between Breaking-Cas and CRISPR Design Tool (MIT) is available here (pdf). Results are very similar yet not identical, probably due to small differences in estimation of pairwise distance between mismatches for each off-target.
External Tools for Evaluating sgRNA Efficiency
Not only the number and characteristics of potential off-targets should
be taken into account when designing sgRNA, but also its on-target
efficiency. Thus, it is advisable to use Breaking-Cas
in conjunction with a tool aimed at predicting on-target efficiency,
since the final goal is to find sgRNAs with high on-target efficiency
and a low number of off-targets.
Several algorithms have been proposed to analyze sgRNA
features that determine efficiency, defined as the capacity
to generate indels at target sites.
As efficiency is specific for each particular Cas nuclease,
algorithms are focused mainly on sgRNAs specific for the
widely used Cas9 and are not generalizable to other systems:
BOX 1: Scores calculations details
A. ESPECIAL CASE: off-target score calculation for Cas9 from Streptococcus pyogenes:
For each off-target, the probability of being a true secondary target for Streptococcus pyogenes's Cas9 is estimated as described in Optimized CRISPR Design tool (Zhang Lab, MIT):
Table 2. Streptococcus pyogenes's Cas9 position-dependent weights for mismatches (crispr.mit.edu/about) |
A score (Soff) is calculated for each off-target based
on the number and position of the mismatches. The higher the score, the higher the
probability of acting as a true secondary Cas9 site. In general, for Streptococcus pyogenes's Cas9 mismatches at last positions
(close to the PAM) strongly decrease the off-target's score. The formula consists of three factors:
T1 x T2 x T3:
The above formula takes into account the influence of the mismatches due to their position (T1), the effect of the mean pairwise distance between mismatches (T2) and penalizes targets with many mismatches (T3). |
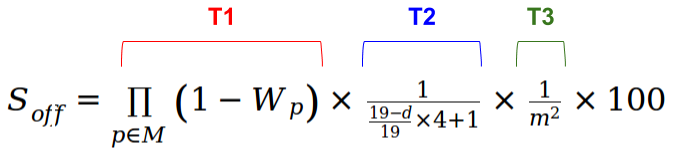

B. GENERAL CASE: off-target score calculation when there is no experimental evidence for mismatch's weights:
If there are no experimental evidence for the positional influence of mismatches, for each off-target, the probability of being a true secondary target is estimated as above except that all positional weights are set to "0" so the factor of the equation that depends on the mismatch's positions (T1) is always 1. Also, in the general case, the "19" in T2 term of the formula is replaced by oligosize-1.
It is possible to introduce customized positional weights (values between 0 and 1, being "0" a totally allowed mismatch and "1" a totally forbiden mismatch) for nucleases with experimental evidences known by the user... modify them at your own risk.
C. Aggregated score for guide candidates:
The sum of all Soff for a candidate guide is used for calculating
a global score (Sguide) as:
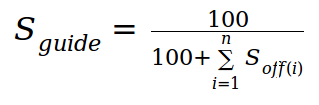
This Sguide can be used as a main criteria to select interesting gRNA candidates: the higher the
aggregate score for a gRNA candidate, the less "problematic" the off-targets will be.
FINAL NOTE:
Oligo alignments with no mismatches (on-targets) are not included in the above formulas and have an imputed score (Soff) of 100.